R for Data Analysis
Overview
Teaching: 150 min
Exercises: 15 minQuestions
How can I summarize my data in R?
How can R help make my research more reproducible?
How can I combine two datasets from different sources?
How can data tidying facilitate answering analysis questions?
Objectives
To become familiar with the functions of the
dplyrandtidyrpackages.To be able to use
dplyrandtidyrto prepare data for analysis.To be able to combine two different data sources using joins.
To be able to create plots and summary tables to answer analysis questions.
Contents
- Getting started
- An introduction to data analysis in R using
dplyr - Cleaning up data
- Joining data frames
- Analyzing combined data
- Finishing with Git and GitHub
Getting Started
First, navigate to the ontario-reports directory however you’d like and open ontario-report.Rproj.
This should open the ontario-report R project in RStudio.
You can check this by seeing if the Files in the bottom right of RStudio are the ones in your ontario-report directory.
Yesterday we spent a lot of time making plots in R using the ggplot2 package. Visualizing data using plots is a very powerful skill in R, but what if we would like to work with only a subset of our data? Or clean up messy data, calculate summary statistics, create a new variable, or join two datasets together? There are several different methods for doing this in R, and we will touch on a few today using functions the dplyr package.
First, we will create a new RScript file for our work. Open RStudio. Choose “File” > “New File” > “RScript”. We will save this file as data_analysis.R
Loading in the data
We will start by importing the complete sample dataset that we used yesterday into our fresh new R session. Today let’s type them into the console ourselves: sample_data <- read_csv("data/sample_data.csv")
Exercise
If we look in the console now, we’ll see we’ve received an error message saying that R “could not find the function
read_csv()”. Hint: Packages…Solution
What this means is that R cannot find the function we are trying to call. The reason for this usually is that we are trying to run a function from a package that we have not yet loaded. This is a very common error message that you will probably see a lot when using R. It’s important to remember that you will need to load any packages you want to use into R each time you start a new session. The
read_csvfunction comes from thereadrpackage which is included in thetidyversepackage so we will just load thetidyversepackage and run the import code again.
Now that we know what’s wrong, We will use the read_csv() function from the Tidyverse readr package. Load the tidyverse package and sample dataset using the code below.
library(tidyverse)
Warning: package 'lubridate' was built under R version 4.3.3
── Attaching core tidyverse packages ─────────────────────────────────────────────────────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.5.1 ✔ tibble 3.2.1
✔ lubridate 1.9.4 ✔ tidyr 1.3.1
✔ purrr 1.0.2
── Conflicts ───────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
The output in your console shows that by doing this, we attach several useful packages for data wrangling, including readr. Check out these packages and their documentation at tidyverse.org
Reminder: Many of these packages, including
dplyr, come with “Cheatsheets” found under the Help RStudio menu tab.
Reload your data:
sample_data <- read_csv("data/sample_data.csv")
Rows: 71 Columns: 9
── Column specification ───────────────────────────────────────────────────────────────────────────────────────────────────────
Delimiter: ","
chr (2): sample_id, env_group
dbl (7): depth, cells_per_ml, temperature, total_nitrogen, total_phosphorus, diss_org_carbon, chlorophyll
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Notice that the output of the read_csv() function is pretty informative. It tells us the name of all of our column headers as well as how it interpreted the data type. This birds-eye-view can help you take a quick look that everything is how we expect it to be.
Now we have the tools necessary to work through this lesson.
An introduction to data analysis in R using dplyr
Get stats fast with summarize()
Let’s say we would like to know what is the mean (average) cell abundance in our dataset. R has a built in function function called mean() that will calculate this value for us. We can apply that function to our cells_per_ml column using the summarize() function. Here’s what that looks like:
summarize(sample_data, average_cells=mean(cells_per_ml))
# A tibble: 1 × 1
average_cells
<dbl>
1 4046023.
When we call summarize(), we can use any of the column names of our data object as values to pass to other functions. summarize() will return a new data object and our value will be returned as a column.
Note: The
summarize()andsummarise()perform identical functions.
We name this new column so we can use in a future argument. So the average_cells= part tells summarize() to use “average_cells” as the name of the new column. Note that you don’t have to quotes around this new name as long as it starts with a letter and doesn’t include a space.
Instead of including the data as an argument, we can use the pipe operator %>% to pass the data value into the summarize function.
sample_data %>% summarize(average_cells=mean(cells_per_ml))
# A tibble: 1 × 1
average_cells
<dbl>
1 4046023.
This line of code will do the exact same thing as our first summary command, but the piping function tells R to use the sample_data dataframe as the first argument in the next function.
This lets us “chain” together multiple functions, which will be helpful later. Note that the pipe (%>%) is a bit different from using the ggplot plus (+). Pipes take the output from the left side and use it as input to the right side. Plusses layer on additional information (right side) to a preexisting plot (left side).
We can also add an
sample_data %>%
summarize(average_cells=mean(cells_per_ml))
# A tibble: 1 × 1
average_cells
<dbl>
1 4046023.
Using the pipe operator %>% and enter command makes our code more readable. The pipe operator %>% also helps to avoid using nested functions and minimizes the need for new variables.
Since we use the pipe operator so often, there is a keyboard shortcut for it in RStudio. You can press
Pro tip: Saving a new dataframe
Notice that when we run the following code, we are not actually saving a new variable:
sample_data %>% summarize(average_cells=mean(cells_per_ml))This simply outputs what we have created, but does not change actually change
sample_dataor save a new dataframe. To save a new dataframe, we could run:sample_data_summarized <- sample_data %>% summarize(average_cells=mean(cells_per_ml))Or if we want to change
sample_dataitself:sample_data <- sample_data %>% summarize(average_cells=mean(cells_per_ml))IMPORTANT: This would overwrite the existing
sample_dataobject. Let’s not do this!For now, we will not be saving dataframes, since we are just experimenting with
dyplrfunctions, but it will be useful later on in this lesson.
Narrow down rows with filter()
Let’s take a look at the value we just calculated, which tells us the average cell abundance for all rows in the data was 4,046,023. However, we previously saw that cell abundance varied widely between our environmental groups. It may be more informative to calculate average cell abundance for each of those groups separately.
Let’s start by just calculating cell abundance within the Deep group. To do that, we will use the filter() function to only use rows for that group before calculating the mean value.
sample_data %>%
filter(env_group == "Deep") %>%
summarize(average=mean(cells_per_ml))
# A tibble: 1 × 1
average
<dbl>
1 1879707.
Filtering the dataset
What is the average chlorophyll concentration in Deep samples? Hint: the column headers identified by
read_csv()showed us there was a column called chlorophyll in the datasetSolution
sample_data %>% filter(env_group == "Deep") %>% summarize(average_chl=mean(chlorophyll))# A tibble: 1 × 1 average_chl <dbl> 1 0.220By combining
filter()andsummarize()we were able to calculate the mean chlorophyll in the Deep samples.What about in shallow September?
Solution
sample_data %>% filter(env_group == "Shallow_September") %>% summarize(average_chl=mean(chlorophyll))# A tibble: 1 × 1 average_chl <dbl> 1 2.09Wow - much higher than in the deep samples!.
Notice how the pipe operator (%>%) allows us to combine these two simple steps into a more complicated data extraction?. We took the data, filtered out the rows, then took the mean value. The argument we pass to filter() needs to be some expression that will return TRUE or FALSE. We can use comparisons like > (greater than) and < (less than) for example. Here we tested for equality using a double equals sign ==. You can also use != to mean “doesn’t equal”. You use == (double equals) when testing if two values are equal, and you use = (single equals) when naming arguments that you are passing to functions. Try changing it to use filter(env_group = "Deep") and see what happens.
Grouping rows using group_by()
We see that the cell abundance in Deep samples is much lower than the value we got using all of the rows. We saw that by using filtering, we can calculate statistics for each group separately, but we need to do this one at a time. Rather that doing a bunch of different filter() statements, we can instead use the group_by() function. The function allows us to tell the code to treat the rows in logical groups, so rather than summarizing over all the rows, we will get one summary value for each group. Here’s what that will look like:
sample_data %>%
group_by(env_group) %>%
summarize(average=mean(cells_per_ml))
# A tibble: 3 × 2
env_group average
<chr> <dbl>
1 Deep 1879707.
2 Shallow_May 4004784.
3 Shallow_September 5906967.
The group_by() function expects you to pass in the name of a column (or multiple columns separated by comma) in your data.
Note that you might get a message about the summarize function regrouping the output by ‘env_group’. This simply indicates what the function is grouping by.
Grouping the data
Try calculating the average temperature by env_group.
Solution
sample_data %>% group_by(env_group) %>% summarize(average=mean(temperature))# A tibble: 3 × 2 env_group average <chr> <dbl> 1 Deep 4.28 2 Shallow_May 7.76 3 Shallow_September 18.3By combining
group_by()andsummarize()we are able to calculate the mean temperature by environmental group.
You can also create more than one new column when you call summarize(). To do so, you must separate your columns with a comma. Building on the code from the last exercise, let’s add a new column that calculates the minimum cell abundance for each env_group.
sample_data %>%
group_by(env_group) %>%
summarize(average=mean(cells_per_ml), min=min(cells_per_ml))
# A tibble: 3 × 3
env_group average min
<chr> <dbl> <dbl>
1 Deep 1879707. 1246414.
2 Shallow_May 4004784. 2300782.
3 Shallow_September 5906967. 3563527.
Make new variables with mutate()
Each time we ran summarize(), we got back fewer rows than passed in. We either got one row back, or one row per group. But sometimes we want to create a new column in our data without changing the number of rows. The function we use to create new columns is called mutate().
We have a column for the total nitrogen and the total phosphorus. Often, we also want to report the ratio between these two nutrients (researchers often report this as a molar ratio, but for simplicity we’ll just use their masses). Here’s what such a mutate() command would look like:
sample_data %>%
mutate(tn_tp_ratio = total_nitrogen / total_phosphorus)
# A tibble: 71 × 10
sample_id env_group depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll tn_tp_ratio
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 May_12_B Deep 103. 2058864. 4.07 465 3.78 2.48 0.05 123.
2 May_12_E Shallow_May 5 4696827. 7.01 465 4.39 2.38 2.53 106.
3 May_12_M Shallow_May 15 4808339. 6.14 474 5.37 2.60 3.2 88.3
4 May_17_E Shallow_May 5 3738681. 5.99 492 4.67 2.44 0.55 105.
5 May_29_B Deep 27 2153086. 4.67 525 4.44 2.40 0.48 118.
6 May_29_E Shallow_May 5 3124920. 5.97 521 3.71 2.28 0.79 140.
7 May_29_M Shallow_May 19 2566156. 5.69 539 4.23 2.33 0.44 127.
8 May_33_B Deep 135 2293177. 3.87 505 4.18 2.34 0.22 121.
9 May_33_E Shallow_May 5 5480859. 7.93 473 6.64 2.51 3.44 71.2
10 May_33_M Shallow_May 20 3114433. 4.53 515 4.14 2.23 1.27 124.
# ℹ 61 more rows
This will add a new column called “tn_tp_ratio” to our data. We use the column names as if they were regular values that we want to perform mathematical operations on and provide the name in front of an equals sign like we have done with summarize()
mutate()We can also multiply by constants or other numbers using mutate - remember how in the plotting lesson we made a plot with cells_per_ml in millions? Try making a new column for this dataframe called cellsInMillions that is the cell abundance in millions.
Solution:
sample_data %>% mutate(tn_tp_ratio = total_nitrogen / total_phosphorus, cellsInMillions = cells_per_ml / 1000000)# A tibble: 71 × 11 sample_id env_group depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll tn_tp_ratio cellsInMillions <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> 1 May_12_B Deep 103. 2058864. 4.07 465 3.78 2.48 0.05 123. 2.06 2 May_12_E Shallow_… 5 4696827. 7.01 465 4.39 2.38 2.53 106. 4.70 3 May_12_M Shallow_… 15 4808339. 6.14 474 5.37 2.60 3.2 88.3 4.81 4 May_17_E Shallow_… 5 3738681. 5.99 492 4.67 2.44 0.55 105. 3.74 5 May_29_B Deep 27 2153086. 4.67 525 4.44 2.40 0.48 118. 2.15 6 May_29_E Shallow_… 5 3124920. 5.97 521 3.71 2.28 0.79 140. 3.12 7 May_29_M Shallow_… 19 2566156. 5.69 539 4.23 2.33 0.44 127. 2.57 8 May_33_B Deep 135 2293177. 3.87 505 4.18 2.34 0.22 121. 2.29 9 May_33_E Shallow_… 5 5480859. 7.93 473 6.64 2.51 3.44 71.2 5.48 10 May_33_M Shallow_… 20 3114433. 4.53 515 4.14 2.23 1.27 124. 3.11 # ℹ 61 more rows
Subset columns using select()
We use the filter() function to choose a subset of the rows from our data, but when we want to choose a subset of columns from our data we use select(). For example, if we only wanted to see the sample IDs (“sample_id”) and depth values, we can do:
sample_data %>%
select(sample_id, depth)
# A tibble: 71 × 2
sample_id depth
<chr> <dbl>
1 May_12_B 103.
2 May_12_E 5
3 May_12_M 15
4 May_17_E 5
5 May_29_B 27
6 May_29_E 5
7 May_29_M 19
8 May_33_B 135
9 May_33_E 5
10 May_33_M 20
# ℹ 61 more rows
We can also use select() to drop/remove particular columns by putting a minus sign (-) in front of the column name. For example, if we want everything but the env_group column, we can do:
sample_data %>%
select(-env_group)
# A tibble: 71 × 8
sample_id depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 May_12_B 103. 2058864. 4.07 465 3.78 2.48 0.05
2 May_12_E 5 4696827. 7.01 465 4.39 2.38 2.53
3 May_12_M 15 4808339. 6.14 474 5.37 2.60 3.2
4 May_17_E 5 3738681. 5.99 492 4.67 2.44 0.55
5 May_29_B 27 2153086. 4.67 525 4.44 2.40 0.48
6 May_29_E 5 3124920. 5.97 521 3.71 2.28 0.79
7 May_29_M 19 2566156. 5.69 539 4.23 2.33 0.44
8 May_33_B 135 2293177. 3.87 505 4.18 2.34 0.22
9 May_33_E 5 5480859. 7.93 473 6.64 2.51 3.44
10 May_33_M 20 3114433. 4.53 515 4.14 2.23 1.27
# ℹ 61 more rows
selecting columns
Create a dataframe with only the
sample_id,env_group,depth,temperature, andcells_per_mlcolumns.Solution:
There are multiple ways to do this exercise. Here are two different possibilities.
sample_data %>% select(sample_id, env_group, depth, temperature, cells_per_ml)# A tibble: 71 × 5 sample_id env_group depth temperature cells_per_ml <chr> <chr> <dbl> <dbl> <dbl> 1 May_12_B Deep 103. 4.07 2058864. 2 May_12_E Shallow_May 5 7.01 4696827. 3 May_12_M Shallow_May 15 6.14 4808339. 4 May_17_E Shallow_May 5 5.99 3738681. 5 May_29_B Deep 27 4.67 2153086. 6 May_29_E Shallow_May 5 5.97 3124920. 7 May_29_M Shallow_May 19 5.69 2566156. 8 May_33_B Deep 135 3.87 2293177. 9 May_33_E Shallow_May 5 7.93 5480859. 10 May_33_M Shallow_May 20 4.53 3114433. # ℹ 61 more rowssample_data %>% select(-total_nitrogen, -total_phosphorus, -diss_org_carbon, -chlorophyll)# A tibble: 71 × 5 sample_id env_group depth cells_per_ml temperature <chr> <chr> <dbl> <dbl> <dbl> 1 May_12_B Deep 103. 2058864. 4.07 2 May_12_E Shallow_May 5 4696827. 7.01 3 May_12_M Shallow_May 15 4808339. 6.14 4 May_17_E Shallow_May 5 3738681. 5.99 5 May_29_B Deep 27 2153086. 4.67 6 May_29_E Shallow_May 5 3124920. 5.97 7 May_29_M Shallow_May 19 2566156. 5.69 8 May_33_B Deep 135 2293177. 3.87 9 May_33_E Shallow_May 5 5480859. 7.93 10 May_33_M Shallow_May 20 3114433. 4.53 # ℹ 61 more rowsDid you notice that the order of columns also changed, to match the order of arguments in the select function?
Pro tip: Selecting a range of columns
Sometimes, we want to select a range of contiguous columns. Rather than writing out each column name, we can use the
:operator to select a range of columnssample_data %>% select(sample_id:temperature)We can also drop a range of columns in this way, though we need to include some extra parentheses:
sample_data %>% select(-(total_nitrogen:chlorophyll))
Bonus: Using helper functions with
select()The
select()function has a bunch of helper functions that are handy if you are working with a dataset that has a lot of columns. You can see these helper functions on the?selecthelp page. For example, let’s say we wanted to select the sample_id column and all the columns that start with “total”. You can do that with:sample_data %>% select(sample_id, starts_with("total"))# A tibble: 71 × 3 sample_id total_nitrogen total_phosphorus <chr> <dbl> <dbl> 1 May_12_B 465 3.78 2 May_12_E 465 4.39 3 May_12_M 474 5.37 4 May_17_E 492 4.67 5 May_29_B 525 4.44 6 May_29_E 521 3.71 7 May_29_M 539 4.23 8 May_33_B 505 4.18 9 May_33_E 473 6.64 10 May_33_M 515 4.14 # ℹ 61 more rowsThis returns just the three columns we are interested in.
Using
select()with a helper functionFind a helper function on the help page that will choose all the columns that have “n” as their last letter
Solution
The helper function
ends_with()can help us here.sample_data %>% select(ends_with("n"))# A tibble: 71 × 2 total_nitrogen diss_org_carbon <dbl> <dbl> 1 465 2.48 2 465 2.38 3 474 2.60 4 492 2.44 5 525 2.40 6 521 2.28 7 539 2.33 8 505 2.34 9 473 2.51 10 515 2.23 # ℹ 61 more rows
Cleaning up data
Researchers are often pulling data from several sources, and the process of making data compatible with one another and prepared for analysis can be a large undertaking. Luckily, there are many functions that allow us to do this in R. We’ve been working with the sample dataset, which contains metadata about each sample we collected. In this section, we practice cleaning and preparing a second dataset derived from sequencing data, which holds the relative abundances of the most abundant Phyla in our dataset.
It’s always good to go into data cleaning with a clear goal in mind. Here, we’d like to prepare the taxon data to be compatible with our sample data so we can directly compare the abundance of specific Phyla to our sample data. To make this work, we’d like a data frame that contains a column with the sample ID, and columns for the abundances of each Phyla. Let’s start with reading the data in using read_csv()
read_csv("data/taxon_abundance.csv")
New names:
• `` -> `...2`
• `` -> `...3`
• `` -> `...4`
• `` -> `...5`
• `` -> `...6`
• `` -> `...7`
• `` -> `...8`
• `` -> `...9`
• `` -> `...10`
Warning: One or more parsing issues, call `problems()` on your data frame for details, e.g.:
dat <- vroom(...)
problems(dat)
Rows: 73 Columns: 10
── Column specification ───────────────────────────────────────────────────────────────────────────────────────────────────────
Delimiter: ","
chr (10): Taxon Abundance from Lake Ontario, ...2, ...3, ...4, ...5, ...6, ...7, ...8, ...9, ...10
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
# A tibble: 73 × 10
`Taxon Abundance from Lake Ontario` ...2 ...3 ...4 ...5 ...6 ...7 ...8 ...9 ...10
<chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
1 Schmidt Lab 2023 <NA> <NA> <NA> <NA> <NA> <NA> <NA> ,
2 sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verr… Cyan… Lot_… <NA> <NA>
3 Sep_43_B 0.4750424903837557 0.14116870918686822 0.072457285982646 0.005154754… 0.11… 0.14… 1720… MiSeq <NA>
4 Sep_29_E 0.4532027397871533 0.18427745922670175 0.08144619709511161 0.006882934… 0.12… 0.11… <NA> MiSeq <NA>
5 Sep_62_B 0.4445189001548922 0.22247366281816092 0.11902538695609188 0.008414715… 0.07… 0.08… <NA> MiSeq <NA>
6 May_8_E 0.44319281443068753 0.1948738307322036 0.2632806490831948 0.052378234… 0.03… 6.94… 1720… MiSeq <NA>
7 Sep_62_E 0.44124389783423684 0.20572502270050613 0.09990919797538457 0.005726820… 0.10… 0.10… <NA> MiSeq <NA>
8 May_38_E 0.4396651527295396 0.17521595867842193 0.3104016386143022 0.016430670… 0.03… 8.01… 1720… MiSeq <NA>
9 Sep_12_E 0.43588136704802166 0.21974786854186404 0.07703854842235662 0.009706332… 0.10… 0.11… 1720… MiSeq <NA>
10 May_17_E 0.4351187991214858 0.19107690632203736 0.21576243805928202 0.084983573… 0.05… 0.00… 1720… MiSeq <NA>
# ℹ 63 more rows
Hmmm, this looks a little weird. It is telling us that our table has 74 rows and 10 columns. What in the world is going on here?
Let’s go to the file in the Files pane and select “View File”.
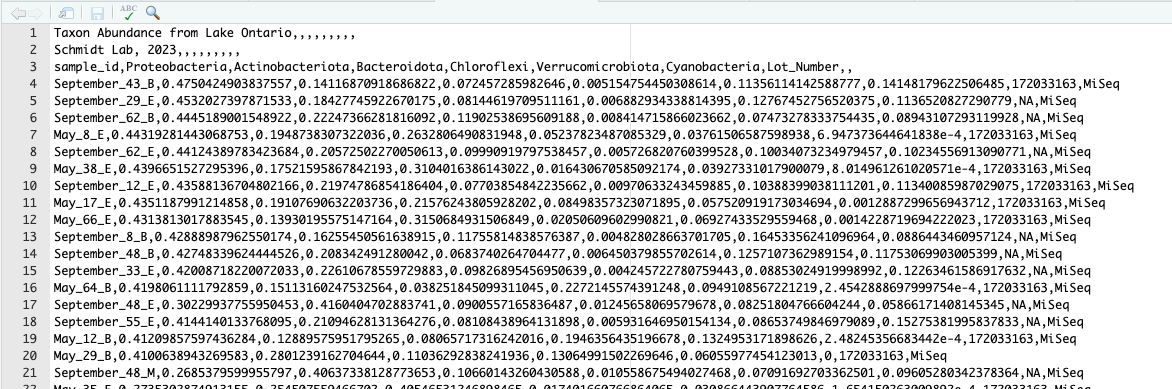
Now, we begin to see at least one of the problems. It looks like there are two descriptive titles at the top of our file, which aren’t part of the data table itself. Ideally, we’d skip those. We can do this using the skip= argument in read_csv by giving it a number of lines to skip.
read_csv("data/taxon_abundance.csv", skip=2)
New names:
• `` -> `...9`
• `` -> `...10`
Warning: One or more parsing issues, call `problems()` on your data frame for details, e.g.:
dat <- vroom(...)
problems(dat)
Rows: 71 Columns: 10
── Column specification ───────────────────────────────────────────────────────────────────────────────────────────────────────
Delimiter: ","
chr (2): sample_id, ...9
dbl (7): Proteobacteria, Actinobacteriota, Bacteroidota, Chloroflexi, Verrucomicrobiota, Cyanobacteria, Lot_Number
lgl (1): ...10
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
# A tibble: 71 × 10
sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verrucomicrobiota Cyanobacteria Lot_Number ...9 ...10
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <lgl>
1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141 172033163 MiSeq NA
2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114 NA MiSeq NA
3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894 NA MiSeq NA
4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695 172033163 MiSeq NA
5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102 NA MiSeq NA
6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801 172033163 MiSeq NA
7 Sep_12_E 0.436 0.220 0.0770 0.00971 0.104 0.113 172033163 MiSeq NA
8 May_17_E 0.435 0.191 0.216 0.0850 0.0575 0.00129 172033163 MiSeq NA
9 May_66_E 0.431 0.139 0.315 0.0205 0.0693 0.00142 172033163 MiSeq NA
10 Sep_8_B 0.429 0.163 0.118 0.00483 0.165 0.0886 NA MiSeq NA
# ℹ 61 more rows
This is working better - we now have 71 rows and 10 columns. However, we’re also getting a message that read_csv has made some new column names. This can happen if your file has data in rows which lack a column name. In that case, dplyr creates new column names, which start with “…”. Here, it looks like the final column contains information about the sequencing machine which generated the data. There is also another column that looks completely empty.
Warnings and Errors
It’s important to differentiate between Warnings and Errors in R. A warning tells us, “you might want to know about this issue, but R still did what you asked”. An error tells us, “there’s something wrong with your code or your data and R didn’t do what you asked”. You need to fix any errors that arise. Warnings, are probably best to resolve or at least understand why they are coming up. {.callout}
Perhaps we should rename this column to something informative, and drop that empty column. We can use the rename function to do so. While we’re at it, let’s save this as an object which we can work with down the line. I’m going to use the glimpse function to see an overview of this table.
taxon_dirty <- read_csv("data/taxon_abundance.csv", skip=2) %>%
select(-...10) %>%
rename(sequencer = ...9)
New names:
• `` -> `...9`
• `` -> `...10`
Warning: One or more parsing issues, call `problems()` on your data frame for details, e.g.:
dat <- vroom(...)
problems(dat)
Rows: 71 Columns: 10
── Column specification ───────────────────────────────────────────────────────────────────────────────────────────────────────
Delimiter: ","
chr (2): sample_id, ...9
dbl (7): Proteobacteria, Actinobacteriota, Bacteroidota, Chloroflexi, Verrucomicrobiota, Cyanobacteria, Lot_Number
lgl (1): ...10
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
head(taxon_dirty, 6)
# A tibble: 6 × 9
sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verrucomicrobiota Cyanobacteria Lot_Number sequencer
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr>
1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141 172033163 MiSeq
2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114 NA MiSeq
3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894 NA MiSeq
4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695 172033163 MiSeq
5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102 NA MiSeq
6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801 172033163 MiSeq
Bonus: Modifying multiple column names at once
Many data analysts prefer to have their column headings and variable names be in all lower case. We can use a variation of
rename(), which isrename_all()that allows us to set all of the column headings to lower case by giving it the name of the tolower function, which makes everything lowercase.read_csv("data/taxon_abundance.csv", skip=2) %>% rename_all(tolower)New names: • `` -> `...9` • `` -> `...10`Warning: One or more parsing issues, call `problems()` on your data frame for details, e.g.: dat <- vroom(...) problems(dat)Rows: 71 Columns: 10 ── Column specification ─────────────────────────────────────────────────────────────────────────────────────────────────────── Delimiter: "," chr (2): sample_id, ...9 dbl (7): Proteobacteria, Actinobacteriota, Bacteroidota, Chloroflexi, Verrucomicrobiota, Cyanobacteria, Lot_Number lgl (1): ...10 ℹ Use `spec()` to retrieve the full column specification for this data. ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.# A tibble: 71 × 10 sample_id proteobacteria actinobacteriota bacteroidota chloroflexi verrucomicrobiota cyanobacteria lot_number ...9 ...10 <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr> <lgl> 1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141 172033163 MiSeq NA 2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114 NA MiSeq NA 3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894 NA MiSeq NA 4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695 172033163 MiSeq NA 5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102 NA MiSeq NA 6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801 172033163 MiSeq NA 7 Sep_12_E 0.436 0.220 0.0770 0.00971 0.104 0.113 172033163 MiSeq NA 8 May_17_E 0.435 0.191 0.216 0.0850 0.0575 0.00129 172033163 MiSeq NA 9 May_66_E 0.431 0.139 0.315 0.0205 0.0693 0.00142 172033163 MiSeq NA 10 Sep_8_B 0.429 0.163 0.118 0.00483 0.165 0.0886 NA MiSeq NA # ℹ 61 more rows
We previously saw how we can subset columns from a data frame using the select function. There are some columns with extraneous information in this dataset. For instance, while information like the sequencer and extraction kit lot_number might be useful to use in the lab, it won’t be a part of our analysis. Let’s subset out the columns we are interested in, which includes the sample IDs and taxon information (columns from Proteobacteria to Cyanobacteria).
Reviewing selecting columns
Select the columns of interest from our dataset. Note, there are multiple ways to do this.
Solution:
taxon_dirty %>% select(sample_id:Cyanobacteria)# A tibble: 71 × 7 sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verrucomicrobiota Cyanobacteria <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> 1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141 2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114 3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894 4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695 5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102 6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801 7 Sep_12_E 0.436 0.220 0.0770 0.00971 0.104 0.113 8 May_17_E 0.435 0.191 0.216 0.0850 0.0575 0.00129 9 May_66_E 0.431 0.139 0.315 0.0205 0.0693 0.00142 10 Sep_8_B 0.429 0.163 0.118 0.00483 0.165 0.0886 # ℹ 61 more rowstaxon_dirty %>% select(-Lot_Number, -sequencer)# A tibble: 71 × 7 sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verrucomicrobiota Cyanobacteria <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> 1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141 2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114 3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894 4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695 5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102 6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801 7 Sep_12_E 0.436 0.220 0.0770 0.00971 0.104 0.113 8 May_17_E 0.435 0.191 0.216 0.0850 0.0575 0.00129 9 May_66_E 0.431 0.139 0.315 0.0205 0.0693 0.00142 10 Sep_8_B 0.429 0.163 0.118 0.00483 0.165 0.0886 # ℹ 61 more rowstaxon_dirty %>% select(sample_id, Proteobacteria, Actinobacteriota, Bacteroidota, Chloroflexi, Verrucomicrobiota, Cyanobacteria)# A tibble: 71 × 7 sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verrucomicrobiota Cyanobacteria <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> 1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141 2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114 3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894 4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695 5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102 6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801 7 Sep_12_E 0.436 0.220 0.0770 0.00971 0.104 0.113 8 May_17_E 0.435 0.191 0.216 0.0850 0.0575 0.00129 9 May_66_E 0.431 0.139 0.315 0.0205 0.0693 0.00142 10 Sep_8_B 0.429 0.163 0.118 0.00483 0.165 0.0886 # ℹ 61 more rows
Let’s go ahead and assign the output of this code chunk, which is the cleaned dataframe, to a variable name:
taxon_clean <- taxon_dirty %>%
select(sample_id:Cyanobacteria)
Looking at your data: You can get a look at your data-cleaning hard work by navigating to the Environment tab in RStudio and clicking the table icon next to the variable name. Notice when we do this, RStudio automatically runs the
View()command. We’ve made a lot of progress! {.callout}
Changing the shape of the data
Data comes in many shapes and sizes, and one way we classify data is either “wide” or “long.” Data that is “long” has one row per observation, and each column represents a unique, independent variable. The sample_data data is in a long format.
In wide data, each row represents a group of observations and each value is placed in a different column rather than a different row. Our taxon data is in wide format, as taxa abundance is spread out across multiple columns which represent different species. To be honest, the lines between “wide” and “long” can be blurry. One clue that your data might be wide is if you have multiple columns which all share the same units and scale, or add up to a larger whole. In our taxon abundance table, values in each row correspond to a fraction of that samples “whole” (100% of its composition).
The tidyr package contains the functions pivot_wider and pivot_longer that make it easy to switch between the two formats. The tidyr package is included in the tidyverse package so we don’t need to do anything to load it. Let’s convert out taxon_clean to long format.
taxon_long <- taxon_clean %>%
pivot_longer(cols = Proteobacteria:Cyanobacteria,
names_to = "Phylum",
values_to = "Abundance")
taxon_long
# A tibble: 426 × 3
sample_id Phylum Abundance
<chr> <chr> <dbl>
1 Sep_43_B Proteobacteria 0.475
2 Sep_43_B Actinobacteriota 0.141
3 Sep_43_B Bacteroidota 0.0725
4 Sep_43_B Chloroflexi 0.00515
5 Sep_43_B Verrucomicrobiota 0.114
6 Sep_43_B Cyanobacteria 0.141
7 Sep_29_E Proteobacteria 0.453
8 Sep_29_E Actinobacteriota 0.184
9 Sep_29_E Bacteroidota 0.0814
10 Sep_29_E Chloroflexi 0.00688
# ℹ 416 more rows
Notice how much longer our table is. We might describe this data as “tidy” because it makes it easy to work with ggplot2 and dplyr functions (this is where the “tidy” in “tidyverse” comes from). For example, this allows us to use group_by to calculate the average relative abundance for each Phylum:
taxon_long %>%
group_by(Phylum) %>%
summarize(avg_abund = mean(Abundance))
# A tibble: 6 × 2
Phylum avg_abund
<chr> <dbl>
1 Actinobacteriota 0.233
2 Bacteroidota 0.160
3 Chloroflexi 0.0805
4 Cyanobacteria 0.0363
5 Proteobacteria 0.334
6 Verrucomicrobiota 0.103
Long format is also, at times, necessary for specific types of plotting. For example, let’s make a very common plot in microbial ecology: the stacked bar plot. Notice how we are piping our dataframe into our ggplot call.
taxon_long %>%
ggplot(aes(x = sample_id, y = Abundance, fill = Phylum)) +
geom_col() +
theme(axis.text.x = element_text(angle = 90))
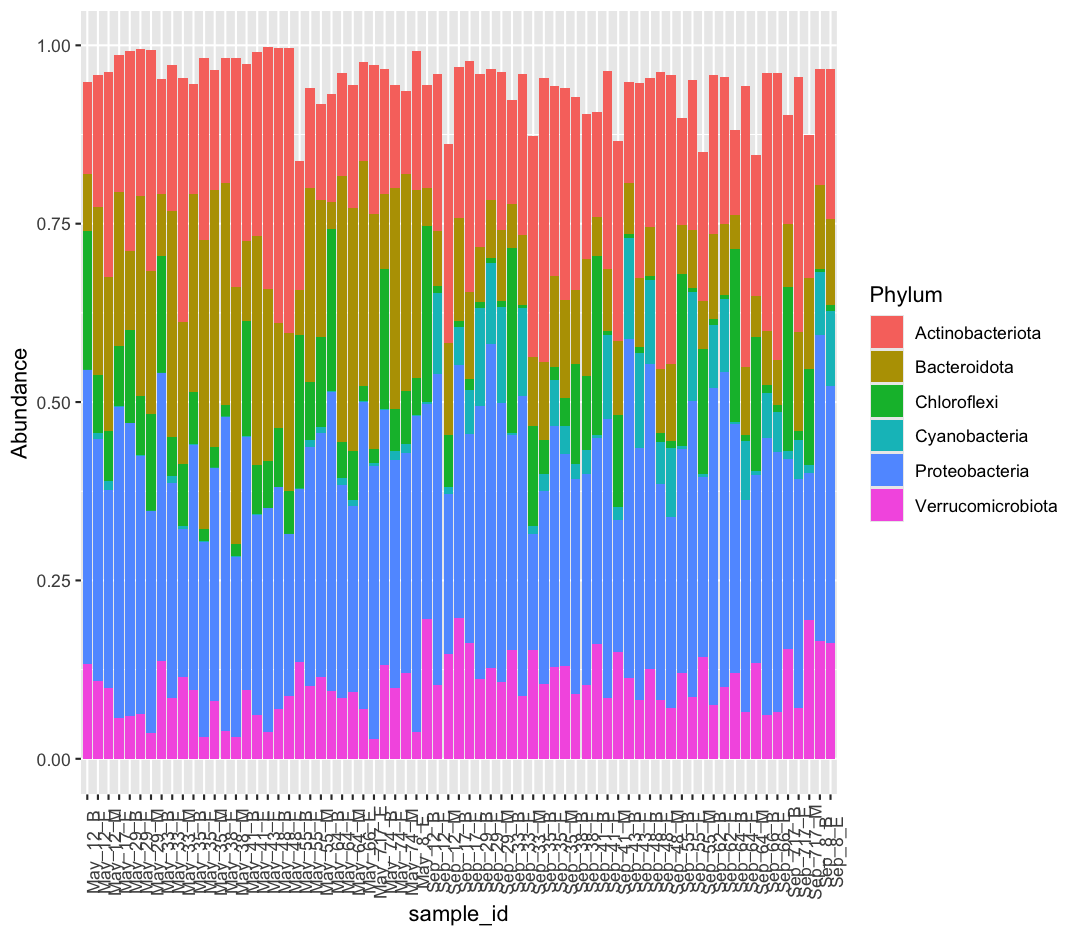
Our bars don’t add up perfectly to one, as our data doesn’t include all Phyla in the original dataset.
If we want to return our data to wide format, we can use pivot_wider
taxon_long %>%
pivot_wider(names_from = "Phylum", values_from = "Abundance")
# A tibble: 71 × 7
sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verrucomicrobiota Cyanobacteria
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141
2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114
3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894
4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695
5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102
6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801
7 Sep_12_E 0.436 0.220 0.0770 0.00971 0.104 0.113
8 May_17_E 0.435 0.191 0.216 0.0850 0.0575 0.00129
9 May_66_E 0.431 0.139 0.315 0.0205 0.0693 0.00142
10 Sep_8_B 0.429 0.163 0.118 0.00483 0.165 0.0886
# ℹ 61 more rows
Joining data frames
Now we’re ready to join our taxon data to the sample data. Previously we saw that we could read in and filter the sample data like this to get the data from the Americas for 2007 so we can create a new dataframe with our filtered data:
head(sample_data, 6)
# A tibble: 6 × 9
sample_id env_group depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 May_12_B Deep 103. 2058864. 4.07 465 3.78 2.48 0.05
2 May_12_E Shallow_May 5 4696827. 7.01 465 4.39 2.38 2.53
3 May_12_M Shallow_May 15 4808339. 6.14 474 5.37 2.60 3.2
4 May_17_E Shallow_May 5 3738681. 5.99 492 4.67 2.44 0.55
5 May_29_B Deep 27 2153086. 4.67 525 4.44 2.40 0.48
6 May_29_E Shallow_May 5 3124920. 5.97 521 3.71 2.28 0.79
head(taxon_clean, 6)
# A tibble: 6 × 7
sample_id Proteobacteria Actinobacteriota Bacteroidota Chloroflexi Verrucomicrobiota Cyanobacteria
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Sep_43_B 0.475 0.141 0.0725 0.00515 0.114 0.141
2 Sep_29_E 0.453 0.184 0.0814 0.00688 0.128 0.114
3 Sep_62_B 0.445 0.222 0.119 0.00841 0.0747 0.0894
4 May_8_E 0.443 0.195 0.263 0.0524 0.0376 0.000695
5 Sep_62_E 0.441 0.206 0.0999 0.00573 0.100 0.102
6 May_38_E 0.440 0.175 0.310 0.0164 0.0393 0.000801
Look at the data in taxon_clean and sample_data. If you had to merge these two data frames together, which column would you use to merge them together? If you said “sample_id” - good job!
We’ll call sample_id our “key”. Now, when we join them together, can you think of any problems we might run into when we merge things? We might not have taxon data for all of the samples in the sample dataset and vice versa.
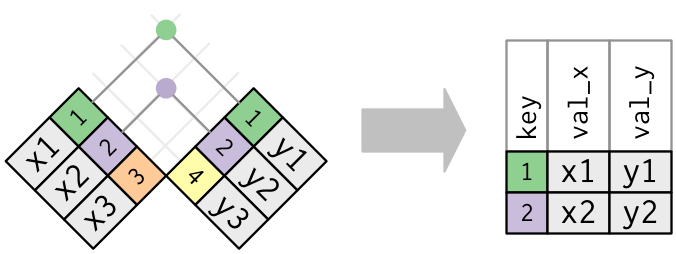
The dplyr package has a number of tools for joining data frames together depending on what we want to do with the rows of the data that are not represented in both data frames. Here we’ll be using inner_join() and anti_join().
In an “inner join”, the new data frame only has those rows where the same key is found in both data frames. This is a very commonly used join.
Bonus: Other dplyr join functions
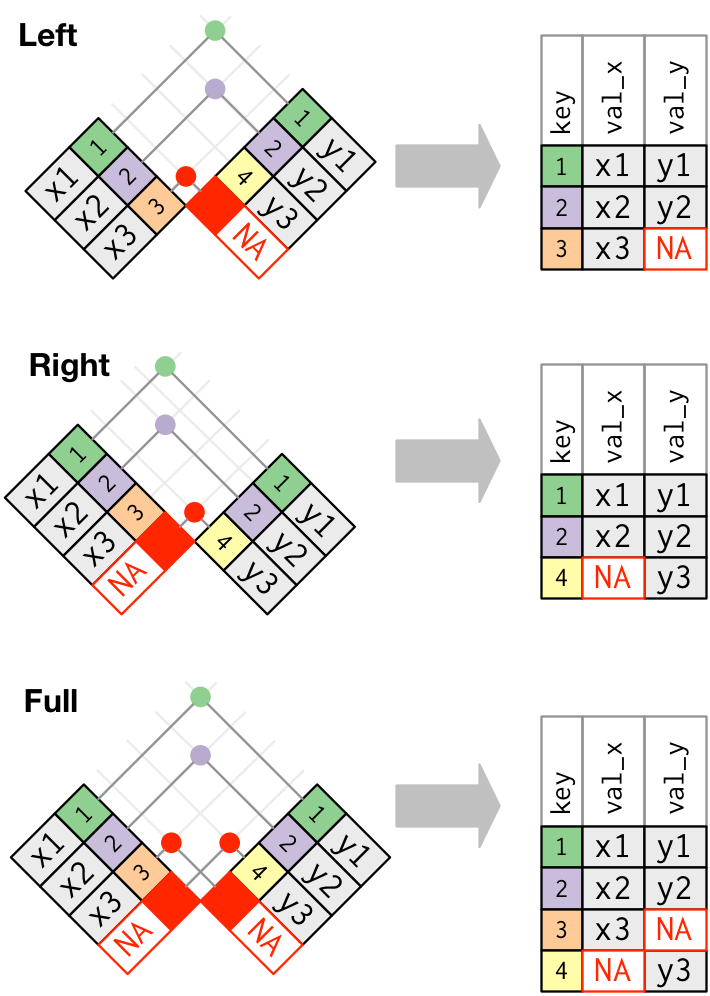
Outer joins and can be performed using
left_join(),right_join(), andfull_join(). In a “left join”, if the key is present in the left hand data frame, it will appear in the output, even if it is not found in the the right hand data frame. For a right join, the opposite is true. For a full join, all possible keys are included in the output data frame.
Let’s give the inner_join() function a try.
inner_join(sample_data, taxon_clean)
Joining with `by = join_by(sample_id)`
# A tibble: 32 × 15
sample_id env_group depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll Proteobacteria
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 May_12_B Deep 103. 2058864. 4.07 465 3.78 2.48 0.05 0.412
2 May_12_E Shallow_May 5 4696827. 7.01 465 4.39 2.38 2.53 0.339
3 May_12_M Shallow_May 15 4808339. 6.14 474 5.37 2.60 3.2 0.276
4 May_17_E Shallow_May 5 3738681. 5.99 492 4.67 2.44 0.55 0.435
5 May_29_B Deep 27 2153086. 4.67 525 4.44 2.40 0.48 0.410
6 May_29_E Shallow_May 5 3124920. 5.97 521 3.71 2.28 0.79 0.362
7 May_29_M Shallow_May 19 2566156. 5.69 539 4.23 2.33 0.44 0.311
8 May_33_B Deep 135 2293177. 3.87 505 4.18 2.34 0.22 0.403
9 May_33_E Shallow_May 5 5480859. 7.93 473 6.64 2.51 3.44 0.301
10 May_33_M Shallow_May 20 3114433. 4.53 515 4.14 2.23 1.27 0.208
# ℹ 22 more rows
# ℹ 5 more variables: Actinobacteriota <dbl>, Bacteroidota <dbl>, Chloroflexi <dbl>, Verrucomicrobiota <dbl>, Cyanobacteria <dbl>
Do you see that we now have data from both data frames joined together in the same data frame? One thing to note about the output is that inner_join() tells us that that it joined by “sample_id”. We can make this explicit using the “by” argument in the join functions
inner_join(sample_data, taxon_clean, by="sample_id")
# A tibble: 32 × 15
sample_id env_group depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll Proteobacteria
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 May_12_B Deep 103. 2058864. 4.07 465 3.78 2.48 0.05 0.412
2 May_12_E Shallow_May 5 4696827. 7.01 465 4.39 2.38 2.53 0.339
3 May_12_M Shallow_May 15 4808339. 6.14 474 5.37 2.60 3.2 0.276
4 May_17_E Shallow_May 5 3738681. 5.99 492 4.67 2.44 0.55 0.435
5 May_29_B Deep 27 2153086. 4.67 525 4.44 2.40 0.48 0.410
6 May_29_E Shallow_May 5 3124920. 5.97 521 3.71 2.28 0.79 0.362
7 May_29_M Shallow_May 19 2566156. 5.69 539 4.23 2.33 0.44 0.311
8 May_33_B Deep 135 2293177. 3.87 505 4.18 2.34 0.22 0.403
9 May_33_E Shallow_May 5 5480859. 7.93 473 6.64 2.51 3.44 0.301
10 May_33_M Shallow_May 20 3114433. 4.53 515 4.14 2.23 1.27 0.208
# ℹ 22 more rows
# ℹ 5 more variables: Actinobacteriota <dbl>, Bacteroidota <dbl>, Chloroflexi <dbl>, Verrucomicrobiota <dbl>, Cyanobacteria <dbl>
One thing to notice is that sample data had 71 rows, but the output of our join only had 32. Let’s investigate. It appears that there must have been samples in the sample data that did not appear in our taxon_clean data frame.
Let’s use anti_join() for this - this will show us the data for the keys on the left that are missing from the data frame on the right.
anti_join(sample_data, taxon_clean, by="sample_id")
# A tibble: 39 × 9
sample_id env_group depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 September_12_B Deep 102 1703592. 4.20 564 1.69 2.42 0.05
2 September_12_E Shallow_September 5 4930739. 18.1 526 3.12 2.96 2.89
3 September_12_M Deep 52 2304545. 4.76 517 2.25 2.58 0.37
4 September_17_B Shallow_September 10.9 6942213. 18.1 385 2.67 2.69 2.28
5 September_17_E Shallow_September 5 7261861. 18.4 396 3.48 2.73 2.74
6 September_29_B Shallow_September 27.9 6168187. 19.0 358 4.28 2.88 2.12
7 September_29_E Shallow_September 5 5582205. 19.0 336 2.68 2.81 2.21
8 September_29_M Shallow_September 15 5681149. 19.0 378 5.08 2.75 2.24
9 September_33_B Deep 135. 1246414. 3.98 506 4.15 2.49 0.05
10 September_33_E Shallow_September 5 7027388. 19.2 370 4.72 2.58 2.5
# ℹ 29 more rows
We can see that none of the joining worked for September samples! What’s going on here? Well, let’s look closer at those sample IDs. If I want to pull just a single column from a datatable, I can use the $ notation
sample_data$sample_id
[1] "May_12_B" "May_12_E" "May_12_M" "May_17_E" "May_29_B" "May_29_E" "May_29_M"
[8] "May_33_B" "May_33_E" "May_33_M" "May_35_B" "May_35_E" "May_35_M" "May_38_E"
[15] "May_38_M" "May_41_B" "May_41_E" "May_43_E" "May_48_B" "May_48_E" "May_55_B"
[22] "May_55_E" "May_55_M" "May_64_B" "May_64_E" "May_64_M" "May_66_E" "May_717_E"
[29] "May_74_B" "May_74_E" "May_74_M" "May_8_E" "September_12_B" "September_12_E" "September_12_M"
[36] "September_17_B" "September_17_E" "September_29_B" "September_29_E" "September_29_M" "September_33_B" "September_33_E"
[43] "September_33_M" "September_35_B" "September_35_E" "September_35_M" "September_38_B" "September_38_E" "September_41_B"
[50] "September_41_E" "September_41_M" "September_43_B" "September_43_E" "September_48_B" "September_48_E" "September_48_M"
[57] "September_55_B" "September_55_E" "September_55_M" "September_62_B" "September_62_E" "September_64_B" "September_64_E"
[64] "September_64_M" "September_66_B" "September_66_E" "September_717_B" "September_717_E" "September_717_M" "September_8_B"
[71] "September_8_E"
taxon_clean$sample_id
[1] "Sep_43_B" "Sep_29_E" "Sep_62_B" "May_8_E" "Sep_62_E" "May_38_E" "Sep_12_E" "May_17_E" "May_66_E" "Sep_8_B" "Sep_48_B"
[12] "Sep_33_E" "May_64_B" "Sep_48_E" "Sep_55_E" "May_12_B" "May_29_B" "Sep_48_M" "May_35_E" "May_33_B" "Sep_66_E" "May_48_E"
[23] "Sep_35_B" "Sep_64_E" "Sep_41_E" "Sep_29_M" "Sep_66_B" "May_48_B" "May_717_E" "Sep_29_B" "May_64_E" "May_29_E" "May_38_M"
[34] "Sep_8_E" "May_35_M" "May_74_B" "Sep_717_E" "Sep_17_B" "May_41_B" "Sep_43_E" "Sep_64_B" "May_35_B" "May_64_M" "May_55_M"
[45] "May_33_M" "May_43_E" "May_12_E" "Sep_35_E" "May_55_E" "Sep_17_E" "May_41_E" "May_74_E" "May_33_E" "Sep_55_B" "May_29_M"
[56] "Sep_33_M" "May_74_M" "Sep_12_B" "Sep_38_B" "Sep_33_B" "Sep_35_M" "Sep_38_E" "Sep_41_B" "May_12_M" "Sep_41_M" "Sep_12_M"
[67] "Sep_717_B" "Sep_64_M" "Sep_55_M" "May_55_B" "Sep_717_M"
In our sample_data, our sample IDs used the full word “September”, but in our taxon table it looks like someone shortened it to “Sep”. As such, inner_join can’t recognize that these are matching keys. Let’s use what we know about mutate alongside a new function, str_replace, to help them match.
taxon_clean_goodSept <- taxon_clean %>%
mutate(sample_id = str_replace(sample_id, pattern = "Sep", replacement = "September"))
Now let’s try with that inner join again.
inner_join(sample_data, taxon_clean_goodSept, by = "sample_id")
# A tibble: 71 × 15
sample_id env_group depth cells_per_ml temperature total_nitrogen total_phosphorus diss_org_carbon chlorophyll Proteobacteria
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 May_12_B Deep 103. 2058864. 4.07 465 3.78 2.48 0.05 0.412
2 May_12_E Shallow_May 5 4696827. 7.01 465 4.39 2.38 2.53 0.339
3 May_12_M Shallow_May 15 4808339. 6.14 474 5.37 2.60 3.2 0.276
4 May_17_E Shallow_May 5 3738681. 5.99 492 4.67 2.44 0.55 0.435
5 May_29_B Deep 27 2153086. 4.67 525 4.44 2.40 0.48 0.410
6 May_29_E Shallow_May 5 3124920. 5.97 521 3.71 2.28 0.79 0.362
7 May_29_M Shallow_May 19 2566156. 5.69 539 4.23 2.33 0.44 0.311
8 May_33_B Deep 135 2293177. 3.87 505 4.18 2.34 0.22 0.403
9 May_33_E Shallow_May 5 5480859. 7.93 473 6.64 2.51 3.44 0.301
10 May_33_M Shallow_May 20 3114433. 4.53 515 4.14 2.23 1.27 0.208
# ℹ 61 more rows
# ℹ 5 more variables: Actinobacteriota <dbl>, Bacteroidota <dbl>, Chloroflexi <dbl>, Verrucomicrobiota <dbl>, Cyanobacteria <dbl>
Woohoo! We matched all of the samples from each table. Let’s save this as a new dataframe, which we can use in later projects.
sample_and_taxon <- inner_join(sample_data, taxon_clean_goodSept, by = "sample_id")
We have reached our data cleaning goals! One of the best aspects of doing all of these steps coded in R is that our efforts are reproducible, and the raw data is maintained. With good documentation of data cleaning and analysis steps, we could easily share our work with another researcher who would be able to repeat what we’ve done. However, it’s also nice to have a saved csv copy of our clean data. That way we can access it later without needing to redo our data cleaning, and we can also share the cleaned data with collaborators. To save our dataframe, we’ll use write_csv().
write_csv(sample_and_taxon, "data/sample_and_taxon.csv")
Great - Now we can move on to the analysis!
Analyzing combined data
Now comes the fun part! All this cleaning and joining has given us a dataset which allows us to look at how environmental variables affects the abundance of different microbial Phyla. For this portion, we are going to focus on the Phylum Chloroflexi. We want to figure out - where do species in this Phylum tend to live in Lake Ontario? Are there differences in its abundance between environmental groups?
To answer the first question, we’ll plot the relative abundance of Chloroflexi against a station’s depth using a scatter plot:
ggplot(sample_and_taxon, aes(x=depth, y=Chloroflexi)) +
geom_point()+
labs(x="Depth (m)",
y="Chloroflexi (Relative Abundance)",
title="There is a strong association between a station's depth \nand the abundance of Chloroflexi"
)
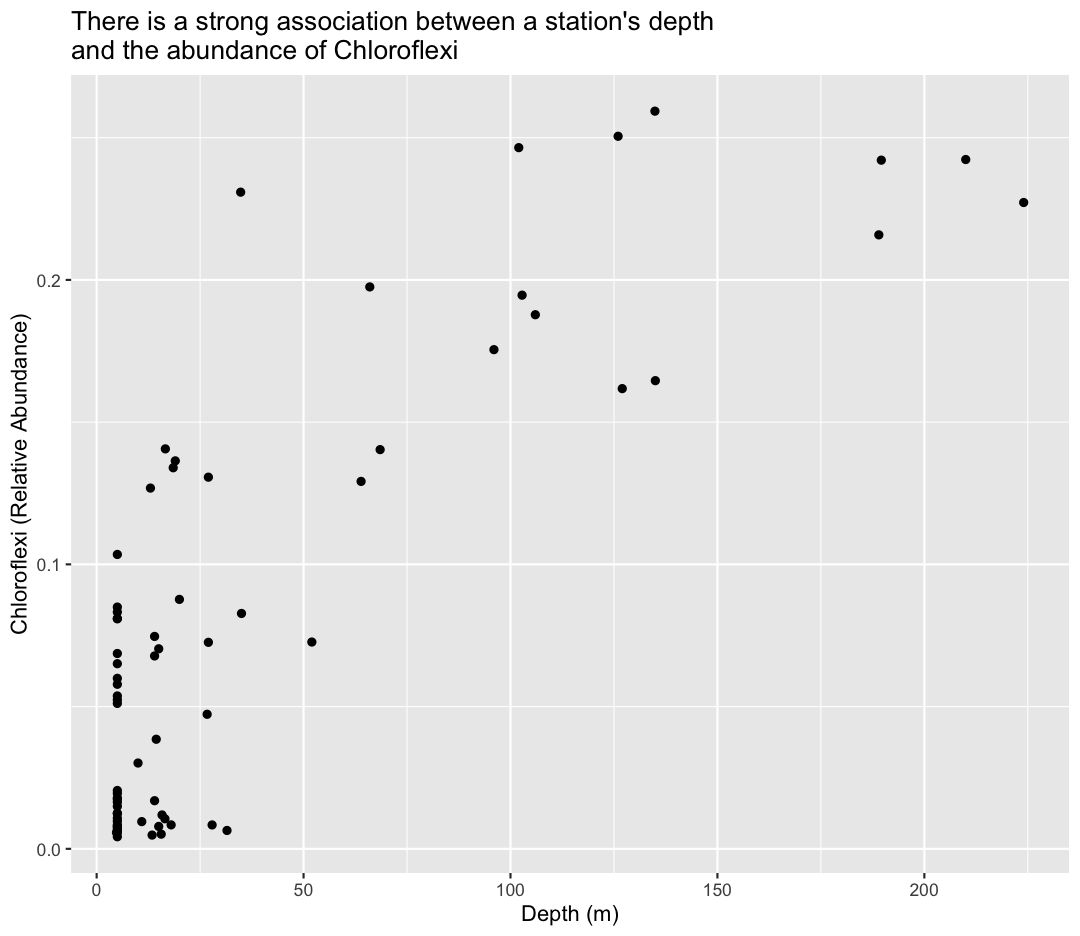
Tip: Notice we used the \n in our title to get a new line to prevent it from getting cut off.
To help clarify the association, we can add a fit line through the data using geom_smooth()
ggplot(sample_and_taxon, aes(x=depth, y=Chloroflexi)) +
geom_point()+
labs(x="Depth (m)",
y="Chloroflexi (Relative Abundance)",
title="There is a strong association between a station's depth \nand the abundance of Chloroflexi"
) +
geom_smooth()
`geom_smooth()` using method = 'loess' and formula = 'y ~ x'
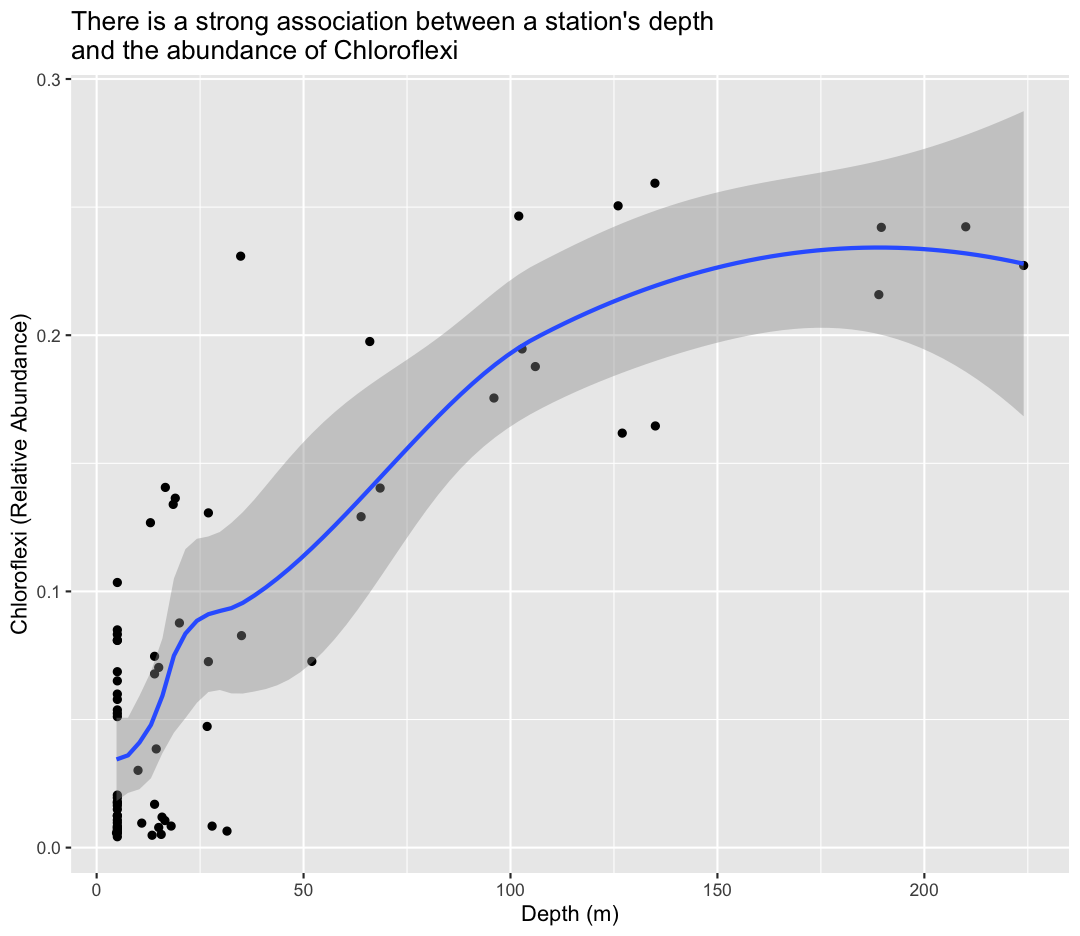
By default, ggplot uses a funky model called “loess estimation” We can force the line to be a linear model (straight) using method="lm" as an argument to geom_smooth
ggplot(sample_and_taxon, aes(x=depth, y=Chloroflexi)) +
geom_point()+
labs(x="Depth (m)",
y="Chloroflexi (Relative Abundance)",
title="There is a strong association between a station's depth \nand the abundance of Chloroflexi"
) +
geom_smooth(method="lm")
`geom_smooth()` using formula = 'y ~ x'
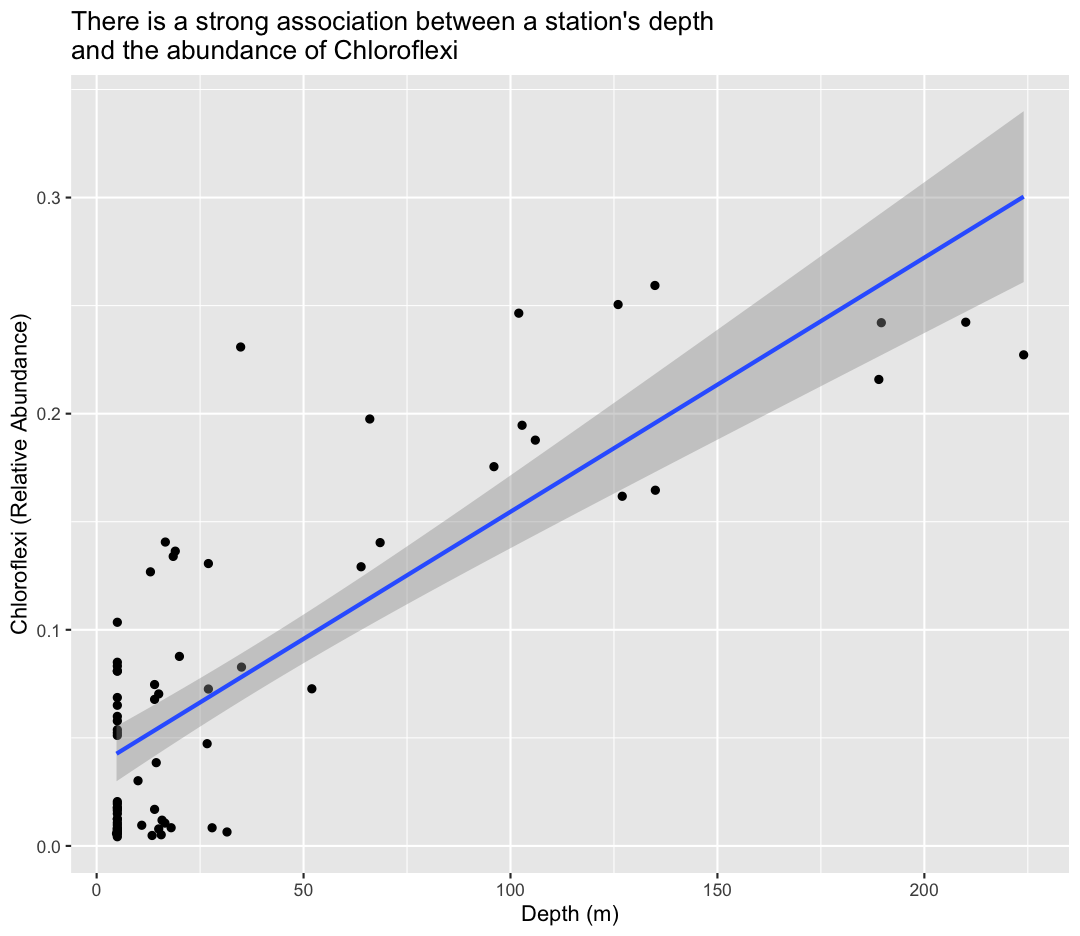
To answer our first question, Chloroflexi appears to be much more abundant in the deeper samples from Lake Ontario!
For the second question, we want to calculate the average abundance of Chloroflexi in our different environmental groups, using a combination of group_by and summarize. Let’s also calculate the standard deviation, to give us a sense of the variance in each group.
sample_and_taxon %>%
group_by(env_group) %>%
summarize(avg_chloro = mean(Chloroflexi),
sd_chloro = sd(Chloroflexi))
# A tibble: 3 × 3
env_group avg_chloro sd_chloro
<chr> <dbl> <dbl>
1 Deep 0.183 0.0539
2 Shallow_May 0.0631 0.0315
3 Shallow_September 0.0116 0.0101
We see that Chloroflexi is most abundant in the Deep groups, relatively rare in Shallow May, and very rare in Shallow September.
Bonus
Bonus content
Sort data with arrange()
The arrange() function allows us to sort our data by some value. Let’s use the sample_data dataframe. We will take the average value for each environmental group and then sort it so the environmental group with the highest cell abundance are on top. Which environmental group might you guess has the highest cell abundance before running the code?
sample_data %>%
group_by(env_group) %>%
summarise(average= mean(cells_per_ml)) %>%
arrange(desc(average))
# A tibble: 3 × 2
env_group average
<chr> <dbl>
1 Shallow_September 5906967.
2 Shallow_May 4004784.
3 Deep 1879707.
Notice there that we can use the column created the in the summarize() step (“average”) later in the arrange() step. We also use the desc() function here to sort the values in a descending order so the largest values are on top. The default is to put the smallest values on top.
Bonus exercises
Calculating absolute abundance
Our data contains total cell abundance (
cells_per_ml) alongside the relative abundance of each Phylum. If we wanted to get the absolute abundance of Chloroflexi (Cells of Chloroflexi per ml), how would we calculate that?Solution
Create a new variable using
mutate()that calculates absolute abundance of Chloroflexi.sample_and_taxon %>% mutate(abs_Chloroflexi = cells_per_ml * Chloroflexi) %>% glimpse()Rows: 71 Columns: 16 $ sample_id <chr> "May_12_B", "May_12_E", "May_12_M", "May_17_E", "May_29_B", "May_29_E", "May_29_M", "May_33_B", "May_33_E", "May_… $ env_group <chr> "Deep", "Shallow_May", "Shallow_May", "Shallow_May", "Deep", "Shallow_May", "Shallow_May", "Deep", "Shallow_May",… $ depth <dbl> 102.8, 5.0, 15.0, 5.0, 27.0, 5.0, 19.0, 135.0, 5.0, 20.0, 27.0, 5.0, 10.0, 5.0, 14.0, 127.0, 5.0, 5.0, 35.0, 5.0,… $ cells_per_ml <dbl> 2058864, 4696827, 4808339, 3738681, 2153086, 3124920, 2566156, 2293177, 5480859, 3114433, 3066162, 5417617, 46103… $ temperature <dbl> 4.07380, 7.01270, 6.13500, 5.99160, 4.66955, 5.97390, 5.68550, 3.87050, 7.93390, 4.53155, 6.57370, 11.22760, 11.0… $ total_nitrogen <dbl> 465, 465, 474, 492, 525, 521, 539, 505, 473, 515, 479, 441, 450, 416, 449, 475, 435, 499, 477, 472, 475, 468, 460… $ total_phosphorus <dbl> 3.78, 4.39, 5.37, 4.67, 4.44, 3.71, 4.23, 4.18, 6.64, 4.14, 4.32, 8.98, 5.99, 8.27, 8.66, 2.52, 5.46, 5.48, 2.31,… $ diss_org_carbon <dbl> 2.478, 2.380, 2.601, 2.435, 2.396, 2.283, 2.334, 2.343, 2.508, 2.232, 2.613, 2.567, 2.628, 2.692, 2.692, 2.389, 2… $ chlorophyll <dbl> 0.05, 2.53, 3.20, 0.55, 0.48, 0.79, 0.44, 0.22, 3.44, 1.27, 1.26, 3.96, 2.44, 3.32, 4.84, 0.21, 2.84, 0.56, 0.24,… $ Proteobacteria <dbl> 0.4120986, 0.3389293, 0.2762080, 0.4351188, 0.4100639, 0.3622527, 0.3110194, 0.4034387, 0.3012852, 0.2077626, 0.3… $ Actinobacteriota <dbl> 0.1288958, 0.1861232, 0.2866884, 0.1910769, 0.2801239, 0.2062492, 0.3100247, 0.1624027, 0.2048393, 0.3409799, 0.1… $ Bacteroidota <dbl> 0.08065717, 0.23470807, 0.21659843, 0.21576244, 0.11036293, 0.27980345, 0.20001474, 0.08615012, 0.31681303, 0.199… $ Chloroflexi <dbl> 0.194635644, 0.080866893, 0.070320609, 0.084983573, 0.130649915, 0.083230440, 0.136388756, 0.164581873, 0.0537443… $ Verrucomicrobiota <dbl> 0.13249532, 0.10878214, 0.09991639, 0.05752092, 0.06055977, 0.06269371, 0.03606823, 0.13660756, 0.08463046, 0.114… $ Cyanobacteria <dbl> 2.482454e-04, 9.574640e-03, 1.262830e-02, 1.288730e-03, 0.000000e+00, 4.535719e-04, 3.315772e-04, 2.399992e-04, 1… $ abs_Chloroflexi <dbl> 400728.37, 379817.78, 338125.36, 317726.48, 281300.48, 260088.49, 349994.76, 377415.41, 294565.42, 273092.26, 222…Previously, we found that Deep samples had the greatest relative abundance of Chloroflexi. Do they also have the greatest absolute abundance of this taxa?
sample_and_taxon %>% mutate(abs_Chloroflexi = cells_per_ml * Chloroflexi) %>% group_by(env_group) %>% summarize(avg_chloro_relative = mean(Chloroflexi), avg_chloro_absolute = mean(abs_Chloroflexi))# A tibble: 3 × 3 env_group avg_chloro_relative avg_chloro_absolute <chr> <dbl> <dbl> 1 Deep 0.183 332349. 2 Shallow_May 0.0631 229210. 3 Shallow_September 0.0116 62915.Looks like yes! However, the difference is much more slight between Deep and Shallow_May when we consider absolute abundance.
Key Points
Package loading is an important first step in preparing an R environment.
Data analsyis in R facilitates reproducible research.
There are many useful functions in the
tidyversepackages that can aid in data analysis.Assessing data source and structure is an important first step in analysis.
Preparing data for analysis can take significant effort and planning.